For me, one of the most annoying features of R is that by default, rbind, cbind and data.frame recycle the shorter vector to the length of the longer vector. I still don’t understand why the standard generics don’t have a parameter like cbind(1:10, 1:5, fill = TRUE) to fill up with ‘NA’s. There may be a deeper reason…
> cbind(1:10, 1:5)
[,1] [,2]
[1,] 1 1
[2,] 2 2
[3,] 3 3
[4,] 4 4
[5,] 5 5
[6,] 6 1
[7,] 7 2
[8,] 8 3
[9,] 9 4
[10,] 10 5
> rbind(1:10, 1:5)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 1 2 3 4 5 6 7 8 9 10
[2,] 1 2 3 4 5 1 2 3 4 5
> data.frame(x = 1:10, y = 1:5)
x y
1 1 1
2 2 2
3 3 3
4 4 4
5 5 5
6 6 1
7 7 2
8 8 3
9 9 4
10 10 5
Even more, using unequal size matrices gives an error:
> mat1 <- matrix(rnorm(10), nrow = 5)
> mat2 <- matrix(rnorm(10), nrow = 2)
> cbind(mat1, mat2)
Fehler in cbind(mat1, mat2) :
Anzahl der Zeilen der Matrizen muss übereinstimmen (siehe Argument 2)
(You can learn some german here. Well, I suppose you know the error…)
Enter cbind.na, rbind.na, data.frame.na !
I modified the three generic functions so that all arguments are filled with ‘NA’s up to maximal occuring length (vector), rows (cbind) or columns (rbind). One must ‘source’ all three functions as cbind.na calls rbind.na etc:
cbind.na <- function (..., deparse.level = 1)
{
na <- nargs() - (!missing(deparse.level))
deparse.level <- as.integer(deparse.level)
stopifnot(0 <= deparse.level, deparse.level <= 2)
argl <- list(...)
while (na > 0 && is.null(argl[[na]])) {
argl <- argl[-na]
na <- na - 1
}
if (na == 0)
return(NULL)
if (na == 1) {
if (isS4(..1))
return(cbind2(..1))
else return(matrix(...)) ##.Internal(cbind(deparse.level, ...)))
}
if (deparse.level) {
symarg <- as.list(sys.call()[-1L])[1L:na]
Nms <- function(i) {
if (is.null(r <- names(symarg[i])) || r == "") {
if (is.symbol(r <- symarg[[i]]) || deparse.level ==
2)
deparse(r)
}
else r
}
}
## deactivated, otherwise no fill in with two arguments
if (na == 0) {
r <- argl[[2]]
fix.na <- FALSE
}
else {
nrs <- unname(lapply(argl, nrow))
iV <- sapply(nrs, is.null)
fix.na <- identical(nrs[(na - 1):na], list(NULL, NULL))
## deactivated, otherwise data will be recycled
#if (fix.na) {
# nr <- max(if (all(iV)) sapply(argl, length) else unlist(nrs[!iV]))
# argl[[na]] <- cbind(rep(argl[[na]], length.out = nr),
# deparse.level = 0)
#}
if (deparse.level) {
if (fix.na)
fix.na <- !is.null(Nna <- Nms(na))
if (!is.null(nmi <- names(argl)))
iV <- iV & (nmi == "")
ii <- if (fix.na)
2:(na - 1)
else 2:na
if (any(iV[ii])) {
for (i in ii[iV[ii]]) if (!is.null(nmi <- Nms(i)))
names(argl)[i] <- nmi
}
}
## filling with NA's to maximum occuring nrows
nRow <- as.numeric(sapply(argl, function(x) NROW(x)))
maxRow <- max(nRow, na.rm = TRUE)
argl <- lapply(argl, function(x) if (is.null(nrow(x))) c(x, rep(NA, maxRow - length(x)))
else rbind.na(x, matrix(, maxRow - nrow(x), ncol(x))))
r <- do.call(cbind, c(argl[-1L], list(deparse.level = deparse.level)))
}
d2 <- dim(r)
r <- cbind2(argl[[1]], r)
if (deparse.level == 0)
return(r)
ism1 <- !is.null(d1 <- dim(..1)) && length(d1) == 2L
ism2 <- !is.null(d2) && length(d2) == 2L && !fix.na
if (ism1 && ism2)
return(r)
Ncol <- function(x) {
d <- dim(x)
if (length(d) == 2L)
d[2L]
else as.integer(length(x) > 0L)
}
nn1 <- !is.null(N1 <- if ((l1 <- Ncol(..1)) && !ism1) Nms(1))
nn2 <- !is.null(N2 <- if (na == 2 && Ncol(..2) && !ism2) Nms(2))
if (nn1 || nn2 || fix.na) {
if (is.null(colnames(r)))
colnames(r) <- rep.int("", ncol(r))
setN <- function(i, nams) colnames(r)[i] <<- if (is.null(nams))
""
else nams
if (nn1)
setN(1, N1)
if (nn2)
setN(1 + l1, N2)
if (fix.na)
setN(ncol(r), Nna)
}
r
}
rbind.na <- function (..., deparse.level = 1)
{
na <- nargs() - (!missing(deparse.level))
deparse.level <- as.integer(deparse.level)
stopifnot(0 <= deparse.level, deparse.level <= 2)
argl <- list(...)
while (na > 0 && is.null(argl[[na]])) {
argl <- argl[-na]
na <- na - 1
}
if (na == 0)
return(NULL)
if (na == 1) {
if (isS4(..1))
return(rbind2(..1))
else return(matrix(..., nrow = 1)) ##.Internal(rbind(deparse.level, ...)))
}
if (deparse.level) {
symarg <- as.list(sys.call()[-1L])[1L:na]
Nms <- function(i) {
if (is.null(r <- names(symarg[i])) || r == "") {
if (is.symbol(r <- symarg[[i]]) || deparse.level ==
2)
deparse(r)
}
else r
}
}
## deactivated, otherwise no fill in with two arguments
if (na == 0) {
r <- argl[[2]]
fix.na <- FALSE
}
else {
nrs <- unname(lapply(argl, ncol))
iV <- sapply(nrs, is.null)
fix.na <- identical(nrs[(na - 1):na], list(NULL, NULL))
## deactivated, otherwise data will be recycled
#if (fix.na) {
# nr <- max(if (all(iV)) sapply(argl, length) else unlist(nrs[!iV]))
# argl[[na]] <- rbind(rep(argl[[na]], length.out = nr),
# deparse.level = 0)
#}
if (deparse.level) {
if (fix.na)
fix.na <- !is.null(Nna <- Nms(na))
if (!is.null(nmi <- names(argl)))
iV <- iV & (nmi == "")
ii <- if (fix.na)
2:(na - 1)
else 2:na
if (any(iV[ii])) {
for (i in ii[iV[ii]]) if (!is.null(nmi <- Nms(i)))
names(argl)[i] <- nmi
}
}
## filling with NA's to maximum occuring ncols
nCol <- as.numeric(sapply(argl, function(x) if (is.null(ncol(x))) length(x)
else ncol(x)))
maxCol <- max(nCol, na.rm = TRUE)
argl <- lapply(argl, function(x) if (is.null(ncol(x))) c(x, rep(NA, maxCol - length(x)))
else cbind(x, matrix(, nrow(x), maxCol - ncol(x))))
## create a common name vector from the
## column names of all 'argl' items
namesVEC <- rep(NA, maxCol)
for (i in 1:length(argl)) {
CN <- colnames(argl[[i]])
m <- !(CN %in% namesVEC)
namesVEC[m] <- CN[m]
}
## make all column names from common 'namesVEC'
for (j in 1:length(argl)) {
if (!is.null(ncol(argl[[j]]))) colnames(argl[[j]]) <- namesVEC
}
r <- do.call(rbind, c(argl[-1L], list(deparse.level = deparse.level)))
}
d2 <- dim(r)
## make all column names from common 'namesVEC'
colnames(r) <- colnames(argl[[1]])
r <- rbind2(argl[[1]], r)
if (deparse.level == 0)
return(r)
ism1 <- !is.null(d1 <- dim(..1)) && length(d1) == 2L
ism2 <- !is.null(d2) && length(d2) == 2L && !fix.na
if (ism1 && ism2)
return(r)
Nrow <- function(x) {
d <- dim(x)
if (length(d) == 2L)
d[1L]
else as.integer(length(x) > 0L)
}
nn1 <- !is.null(N1 <- if ((l1 <- Nrow(..1)) && !ism1) Nms(1))
nn2 <- !is.null(N2 <- if (na == 2 && Nrow(..2) && !ism2) Nms(2))
if (nn1 || nn2 || fix.na) {
if (is.null(rownames(r)))
rownames(r) <- rep.int("", nrow(r))
setN <- function(i, nams) rownames(r)[i] <<- if (is.null(nams))
""
else nams
if (nn1)
setN(1, N1)
if (nn2)
setN(1 + l1, N2)
if (fix.na)
setN(nrow(r), Nna)
}
r
}
data.frame.na <- function (..., row.names = NULL, check.rows = FALSE, check.names = TRUE,
stringsAsFactors = FALSE)
{
data.row.names <- if (check.rows && is.null(row.names))
function(current, new, i) {
if (is.character(current))
new <- as.character(new)
if (is.character(new))
current <- as.character(current)
if (anyDuplicated(new))
return(current)
if (is.null(current))
return(new)
if (all(current == new) || all(current == ""))
return(new)
stop(gettextf("mismatch of row names in arguments of 'data.frame', item %d",
i), domain = NA)
}
else function(current, new, i) {
if (is.null(current)) {
if (anyDuplicated(new)) {
warning("some row.names duplicated: ", paste(which(duplicated(new)),
collapse = ","), " --> row.names NOT used")
current
}
else new
}
else current
}
object <- as.list(substitute(list(...)))[-1L]
mrn <- is.null(row.names)
x <- list(...)
n <- length(x)
if (n < 1L) {
if (!mrn) {
if (is.object(row.names) || !is.integer(row.names))
row.names <- as.character(row.names)
if (any(is.na(row.names)))
stop("row names contain missing values")
if (anyDuplicated(row.names))
stop("duplicate row.names: ", paste(unique(row.names[duplicated(row.names)]),
collapse = ", "))
}
else row.names <- integer(0L)
return(structure(list(), names = character(0L), row.names = row.names,
class = "data.frame"))
}
vnames <- names(x)
if (length(vnames) != n)
vnames <- character(n)
no.vn <- !nzchar(vnames)
vlist <- vnames <- as.list(vnames)
nrows <- ncols <- integer(n)
for (i in seq_len(n)) {
xi <- if (is.character(x[[i]]) || is.list(x[[i]]))
as.data.frame(x[[i]], optional = TRUE, stringsAsFactors = stringsAsFactors)
else as.data.frame(x[[i]], optional = TRUE)
nrows[i] <- .row_names_info(xi)
ncols[i] <- length(xi)
namesi <- names(xi)
if (ncols[i] > 1L) {
if (length(namesi) == 0L)
namesi <- seq_len(ncols[i])
if (no.vn[i])
vnames[[i]] <- namesi
else vnames[[i]] <- paste(vnames[[i]], namesi, sep = ".")
}
else {
if (length(namesi))
vnames[[i]] <- namesi
else if (no.vn[[i]]) {
tmpname <- deparse(object[[i]])[1L]
if (substr(tmpname, 1L, 2L) == "I(") {
ntmpn <- nchar(tmpname, "c")
if (substr(tmpname, ntmpn, ntmpn) == ")")
tmpname <- substr(tmpname, 3L, ntmpn - 1L)
}
vnames[[i]] <- tmpname
}
}
if (missing(row.names) && nrows[i] > 0L) {
rowsi <- attr(xi, "row.names")
nc <- nchar(rowsi, allowNA = FALSE)
nc <- nc[!is.na(nc)]
if (length(nc) && any(nc))
row.names <- data.row.names(row.names, rowsi,
i)
}
nrows[i] <- abs(nrows[i])
vlist[[i]] <- xi
}
nr <- max(nrows)
for (i in seq_len(n)[nrows < nr]) {
xi <- vlist[[i]]
if (nrows[i] > 0L) {
xi <- unclass(xi)
fixed <- TRUE
for (j in seq_along(xi)) {
### added NA fill to max length/nrow
xi1 <- xi[[j]]
if (is.vector(xi1) || is.factor(xi1))
xi[[j]] <- c(xi1, rep(NA, nr - nrows[i]))
else if (is.character(xi1) && class(xi1) == "AsIs")
xi[[j]] <- structure(c(xi1, rep(NA, nr - nrows[i])),
class = class(xi1))
else if (inherits(xi1, "Date") || inherits(xi1,
"POSIXct"))
xi[[j]] <- c(xi1, rep(NA, nr - nrows[i]))
else {
fixed <- FALSE
break
}
}
if (fixed) {
vlist[[i]] <- xi
next
}
}
stop("arguments imply differing number of rows: ", paste(unique(nrows),
collapse = ", "))
}
value <- unlist(vlist, recursive = FALSE, use.names = FALSE)
vnames <- unlist(vnames[ncols > 0L])
noname <- !nzchar(vnames)
if (any(noname))
vnames[noname] <- paste("Var", seq_along(vnames), sep = ".")[noname]
if (check.names)
vnames <- make.names(vnames, unique = TRUE)
names(value) <- vnames
if (!mrn) {
if (length(row.names) == 1L && nr != 1L) {
if (is.character(row.names))
row.names <- match(row.names, vnames, 0L)
if (length(row.names) != 1L || row.names < 1L ||
row.names > length(vnames))
stop("row.names should specify one of the variables")
i <- row.names
row.names <- value[[i]]
value <- value[-i]
}
else if (!is.null(row.names) && length(row.names) !=
nr)
stop("row names supplied are of the wrong length")
}
else if (!is.null(row.names) && length(row.names) != nr) {
warning("row names were found from a short variable and have been discarded")
row.names <- NULL
}
if (is.null(row.names))
row.names <- .set_row_names(nr)
else {
if (is.object(row.names) || !is.integer(row.names))
row.names <- as.character(row.names)
if (any(is.na(row.names)))
stop("row names contain missing values")
if (anyDuplicated(row.names))
stop("duplicate row.names: ", paste(unique(row.names[duplicated(row.names)]),
collapse = ", "))
}
attr(value, "row.names") <- row.names
attr(value, "class") <- "data.frame"
value
}
So, let’s start!
> cbind.na(1:8, 1:7, 1:5, 1:10)
[,1] [,2] [,3] [,4]
[1,] 1 1 1 1
[2,] 2 2 2 2
[3,] 3 3 3 3
[4,] 4 4 4 4
[5,] 5 5 5 5
[6,] 6 6 NA 6
[7,] 7 7 NA 7
[8,] 8 NA NA 8
[9,] NA NA NA 9
[10,] NA NA NA 10
> rbind.na(1:8, 1:7, 1:5, 1:10)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 1 2 3 4 5 6 7 8 NA NA
[2,] 1 2 3 4 5 6 7 NA NA NA
[3,] 1 2 3 4 5 NA NA NA NA NA
[4,] 1 2 3 4 5 6 7 8 9 10
> data.frame.na(A = 1:7, B = 1:5, C = letters[1:3],
+ D = factor(c(1, 1, 2, 2)))
A B C D
1 1 1 a 1
2 2 2 b 1
3 3 3 c 2
4 4 4 <NA> 2
5 5 5 <NA> NA
6 6 NA <NA> NA
7 7 NA <NA> NA
System time overhead for NA-filling is not too much:
> testList <- vector("list", length = 10000)
> for (i in 1:10000) testList[[i]] <- sample(1:100)
> system.time(res1 <- do.call(cbind, testList))
user system elapsed
0.01 0.00 0.02
> testList <- vector("list", length = 10000)
> for (i in 1:10000) testList[[i]] <- sample(1:100, sample(1:100))
> system.time(res2 <- do.call(cbind.na, testList))
user system elapsed
0.23 0.00 0.24
Have fun!





 Posted by anspiess
Posted by anspiess 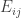 of matrix
of matrix 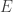 . Consider this:
. Consider this: are distributed normally. In case of heteroscedastic data (i.e. when the variance is dependent on the magnitude of the data), weighting the fit is essential. In nls (or nlsLM of the minpack.lm package), weighting can be conducted by two different methods:
are distributed normally. In case of heteroscedastic data (i.e. when the variance is dependent on the magnitude of the data), weighting the fit is essential. In nls (or nlsLM of the minpack.lm package), weighting can be conducted by two different methods: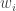 for every
for every  . 2) is a bit more cumbersome and one does not always want to define a new weighted version of the objective function.
. 2) is a bit more cumbersome and one does not always want to define a new weighted version of the objective function. exist, the error
exist, the error  ,
, of the model,
of the model, of the model,
of the model, :
: :
: :
: :
: